

小麦等大基因组作物核心基因组低成本组装及新基因挖掘研究获进展
来源:《核酸研究》
作者:齐美芳等
时间:2018-07-11
6 月 21 日,Nucleic Acids Research 期刊在线发表中国科学院分子植物科学卓越创新中心/植物生理生态研究所张一婧研究组与中科院遗传与发育生物学研究所童依平研究组合作完成的题为 CGT-seq: epigenome-guided de novo assembly of the core genome for divergent populations with large genome 的方法学论文。该工作开发并优化实验与计算流程,实现低成本组装小麦等大基因组作物的核心基因组。
博士研究生齐美芳、李子娟和刘春梅为共同第一作者,水稻实验材料及数据获得植生生态所研究员林鸿宣的帮助。相关工作得到中科院 A 类先导及自然科学基因项目的资助。
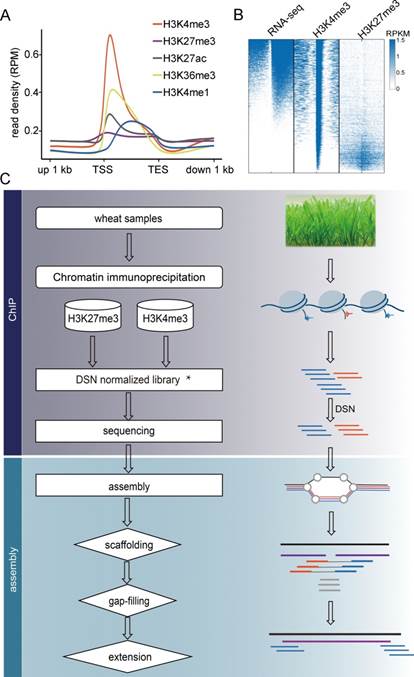
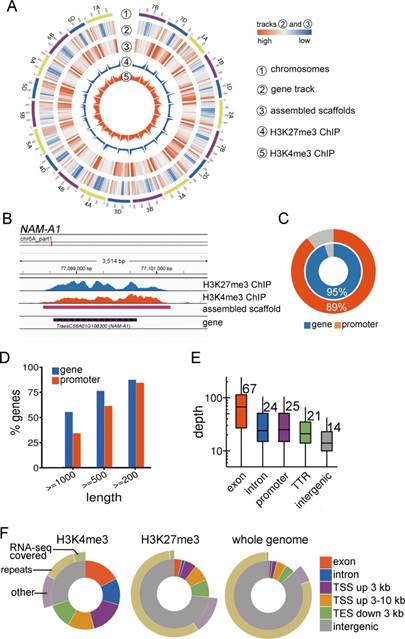
植物高度的遗传多态性为分子育种提供了丰富的遗传资源,确定重要农艺性状的根本方法在于比较不同群体或比较栽培种和野生种间遗传多态性与表型的关联。然而,很多经济物种经历了长期的驯化,基因组复杂而庞大。例如,目前普遍种植的小麦是 6 倍体,全基因组有 17Gb,另外,广泛栽培的大麦、棉花、玉米、花生和大豆都具有 Gb 尺度的基因组,即便是覆盖度要求较低的重测序实验都需要极高的成本。而且,还存在不少未测序的大基因组经济物种,全基因组测序成本非常高,特别是对于群体水平的研究全基因组测序不现实。怎样有效刻画大基因组多态性群体的遗传多样性是一个挑战性的工作。由于很多研究并不需要知道基因组所有的碱基序列,所以人们针对大基因组物种开发了各种低成本的替代测序技术。其基本原理通常是对全基因组序列进行选择性测序,但是这些方法普遍对已有的基因组序列信息要求高,而对于遗传变异大的群体,依赖参考基因组的技术,包括外显子测序,甚至全基因组重测序,都会显著低估多态性。因而,开发不依赖参考基因组直接捕获基因及调控区序列的简化基因组测序方法对于研究多态性高的群体具有重要价值。该方法的理论依据在于调控基因活性的重要表观修饰普遍富集在基因及启动子区(图 A -B),通过免疫共沉淀技术及优化拼接方案从而有效获得基因及附近序列(图 C)。对小麦中国春品种进行核心基因组组装获得的片段与基因区域高度吻合(图 D),能够高效挖掘新基因(图 E -F)、调控区域(图 G)及多态性位点(图 H -J)。该方法已申请专利,其优势在于不依赖参考基因组序列,直接捕获基因及调控区序列,从而极大地降低群体核心基因组拼接的成本,有力地提高大基因组物种的分子遗传与群体遗传学研究效率。(来源:上海生命科学研究院)
CGT-seq: epigenome-guided de novo assembly of the core genome for divergent populations with large genome
Abstract Genetic diversity in plants is remarkably high. Recent whole genome sequencing (WGS) of 67 rice accessions recovered 10,872 novel genes. Comparison of the genetic architecture among divergent populations or between crops and wild relatives is essential for obtaining functional components determining crucial traits. However, many major crops have gigabase-scale genomes, which are not well-suited to WGS. Existing cost-effective sequencing approaches including re-sequencing, exome-sequencing and restriction enzyme-based methods all have difficulty in obtaining long novel genomic sequences from highly divergent population with large genome size. The present study presented a reference-independent core genome targeted sequencing approach, CGT-seq, which employed epigenomic information from both active and repressive epigenetic marks to guide the assembly of the core genome mainly composed of promoter and intragenic regions. This method was relatively easily implemented, and displayed high sensitivity and specificity for capturing the core genome of bread wheat. 95% intragenic and 89% promoter region from wheat were covered by CGT-seq read. We further demonstrated in rice that CGT-seq captured hundreds of novel genes and regulatory sequences from a previously unsequenced ecotype. Together, with specific enrichment and sequencing of regions within and nearby genes, CGT-seq is a time- and resource-effective approach to profiling functionally relevant regions in sequenced and non-sequenced populations with large genomes.
原文链接:https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gky522/5042041
