

药用植物丹参种子的核心微生物群
来源:《International Journal of Molecular Sciences》
作者:陈海敏等
时间:2018-05-04
种子生产是植物生命史上最重要的阶段之一。种子拥有高度多样性的微生物类群,被称为种子相关微生物群,它是与种子相关的内生或附生微生物群落。种子微生物可以允许跨世代的垂直传播,并且对植物生态,健康和生产力有深远影响。
核心微生物组的概念首先为人类微生物组建立,并进一步扩展到其他宿主相关的微生物组如植物等。几种模式植物,如拟南芥,玉米,水稻,大麦和大豆已经实现了植物核心微生物组的组成和功能分析。人类微生物组学研究表明,人类微生物群落的联合对宿主代谢产生巨大影响,但很少有研究分析植物微生物对宿主代谢的影响。现有的植物微生物组研究集中在根际和叶周微生物群落,而对种子微生物组的理解仍然有限。
丹参是重要的药用植物,主要用于治疗冠心病和脑血管疾病。丹参酮和丹酚酸是丹参的两个重要活性成分。几项关于丹参相关微生物的研究主要集中在内生菌和菌根真菌的体外活性上,一些内生真菌可以在宿主植物中产生相似的活性成分。然而,丹参中根,叶或种子相关微生物的组成和功能还没有被破译。
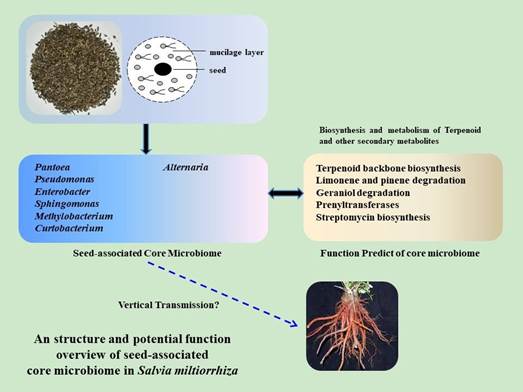
本研究收集了不同地理种植区域的不同种子,并通过高通量测序的方法对与种子相关的微生物组进行了特征分析,以破译丹参中与种子相关的微生物组。从陕西,山西,河南和山东等丹参的主要种植地区进行采样,共7个采样点,每个地理来源的种子收集3个独立重复,并选取一种丹参变种Bge. f作为对照。利用16S rRNA和ITS测序技术,分析不同来源种子和它们共同的微生物分类群之间的重叠。此外,研究还分析了丹参,玉米,豆类,水稻和十字花科微生物分类群的重叠情况。最后,通过PICRUSt软件预测了丹参种子核心微生物组的细菌功能谱。
利用16S rDNA和ITS测序分析了7个不同地理来源的21个丹参种子的核心微生物组,随后进行了生物信息学分析。结果发现整个细菌微生物组属于17个门、39个纲。属于核心细菌微生物组的有γ变形菌(67.6%),α变形菌(15.6%),β变形菌(2.6%),鞘脂杆菌(5.0%),杆菌(4.6%)和放线菌(2.9%)。在丹参种子中,座囊菌纲占核心真菌微生物组的94%,另外两个优势菌群是锤舌菌纲(3.0%)和银耳纲(2.0%)。利用PICRUSt软件进行功能预测发现萜类骨架的生物合成,苎烯,蒎烯和香叶醇和异戊烯转移酶的降解等在核心细菌微生物群中占比较高。研究还发现细菌中的泛菌属,假单胞菌属和鞘氨醇单胞菌属是丰富的核心类群,并且在丹参,玉米,豆类和水稻中是重叠的,而真菌中的链格孢属则在丹参,豆类和十字花科中共享。这些发现强调种子相关微生物是植物微生物组的重要组成部分,可能是药用植物次生代谢的基因库。
本研究中的PICRUSt预测分析表明,这些微生物核心分类群可以影响丹参的生长和质量。本研究发现种子相关的微生物组可能是除宿主植物基因组外的一个储存库和次生代谢能力的补充,种子微生物组的核心分类群不仅促进种子萌发和植物生长,而且调控和参与寄主植物的次生代谢。
Core Microbiome of Medicinal Plant Salvia miltiorrhiza Seed: A Rich Reservoir of Beneficial Microbes for Secondary Metabolism
Abstract Seed microbiome includes special endophytic or epiphytic microbial taxa associated with seeds, which affects seed germination, plant growth, and health. Here, we analyzed the core microbiome of 21Salvia miltiorrhizaseeds from seven different geographic origins using 16S rDNA and ITS amplicon sequencing, followed by bioinformatics analysis. The whole bacterial microbiome was classified into 17 microbial phyla and 39 classes. Gammaproteobacteria (67.6%), Alphaproteobacteria (15.6%), Betaproteobacteria (2.6%), Sphingobacteria (5.0%), Bacilli (4.6%), and Actinobacteria (2.9%) belonged to the core bacterial microbiome. Dothideomycetes comprised 94% of core fungal microbiome inS. miltiorrhizaseeds, and another two dominant classes were Leotiomycetes (3.0%) and Tremellomycetes (2.0%). We found that terpenoid backbone biosynthesis, degradation of limonene, pinene, and geraniol, and prenyltransferases, were overrepresented in the core bacterial microbiome using phylogenetic examination of communities by reconstruction of unobserved states (PICRUSt) software. We also found that the bacterial generaPantoea, Pseudomonas, andSphingomonaswere enriched core taxa and overlapped amongS. miltiorrhiza, maize, bean, and rice, while a fungal genus,Alternaria, was shared withinS. miltiorrhiza, bean, and Brassicaceae families. These findings highlight that seed-associated microbiomeis an important component of plant microbiomes, which may be a gene reservoir for secondary metabolism in medicinal plants.
原文链接:http://www.mdpi.com/1422-0067/19/3/672
