

两项研究发现细胞死亡操控分子的新作用
来源:《Cell》
作者:Christopher P. Dillon等
时间:2014-05-21
肿瘤坏死因子(Tumor necrosis factor,TNF)根据接收的环境信号,刺激细胞死亡或存活。在这个过程中,有两个关键的蛋白:受体相互作用蛋白1(RIP1)和受体相互作用蛋白3(RIP3),形成复合物的形式激发坏死。研究显示,这两种蛋白及其激酶参与触发了细胞死亡和炎症过程,造成许多疾病中的严重后果。
近期两个研究组围绕受体相互作用蛋白激酶 RIPK1 和 RIPK3 展开了研究,发现前者能阻断 Caspase-8 和 RIPK3 介导的一种致命性死亡,另外 RIPK1 也在细胞决定生或死,以及选择如何死去过程中起至关重要的作用。
之前的研究表明, RIPK1 参与了依赖或不依赖 RIPK-3 的信号通路,这些信号通路决定了细胞的死亡及炎症反应。遗传敲除 RIPK1 基因会引发出生后致死,但是缺失 RIPK1 , RIPK3 ,或者caspase- 8, FADD 蛋白的动物却能存活下来,甚至生长至成熟个体。这颇为令人感到疑惑,对此研究人员展开了深入探索,发现 RIPK1 能阻断 Caspase-8 和 RIPK3 介导的出生后早期死亡。
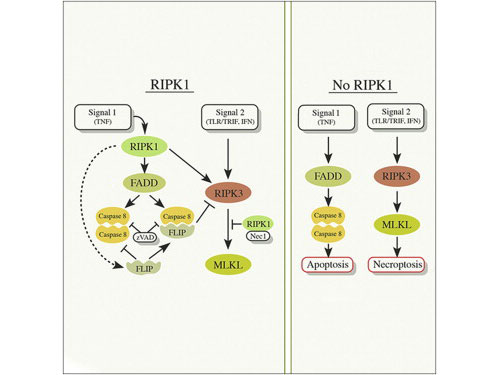
体外实验表明 RIPK1 能限制 Caspase-8 依赖性, TNFR 诱导的细胞凋亡,但缺失 RIPK1 , RIPK3 和 TNFR 1的动物能存活至成年,研究人员还发现 RIPK3 能增加 RIPK1 −/−小鼠的致死率,这说明前者的活性受到了 RIPK1 的抑制。而 TNFR 诱导的 RIPK3 依赖性细胞坏死需要 RIPK1 ,如果细胞缺少 RIPK1 ,就更易发生由 poly:C 或干扰素引发的细胞坏死。
这些研究结果揭示了 RIPK1 在出生后小鼠中的复杂作用,也为进一步了解 RIPK1 相关的 FADD-Caspase-8 , RIPK3 -MLKL信号通路调控提供了新的数据资料。
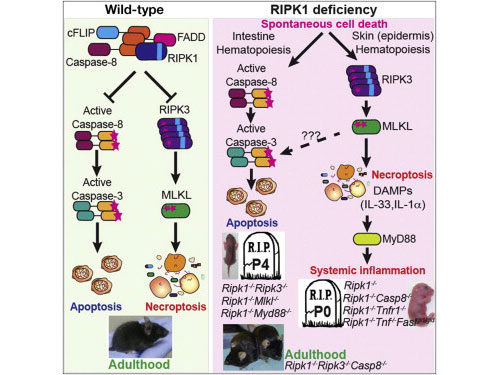
另外一篇文章则发现体内 RIPK1 不仅是启动坏死性凋亡的必要条件,对于抑制坏死性凋亡以及可引起严重组织损伤的失控性炎症也至关重要。
坏死性凋亡是一种“受控”的死亡类型,它在向细胞下达死亡指令的同时,刺激了一种炎症反应让免疫系统知道出现了一些问题。然而,当这一细胞死亡信号通路开始出现失控时,它可以导致炎症性疾病。坏死性凋亡还牵涉到神经退行性疾病、失血引起的脑损伤,以及某些病毒感染。
研究人员不仅证实 RIPK1 对抑制坏死性凋亡至关重要,而且也发现 RIPK1 对骨髓移植后维持造血干细胞的存活起至关重要的作用。考虑到 RIPK1 靶向治疗这一研究发现尤其的重要,因为其有可能会对机体的其他细胞造成不必要的副作用。因此,确保严格地调查所有潜在药物的脱靶效应非常的重要。(来源:360生物谷)
RIPK1 Blocks Early Postnatal Lethality Mediated by Caspase-8 and RIPK3
Summary Receptor-interacting protein kinase (RIPK)-1 is involved in RIPK3-dependent and -independent signaling pathways leading to cell death and/or inflammation. Genetic ablation of ripk1 causes postnatal lethality, which was not prevented by deletion of ripk3, caspase-8, or fadd. However, animals that lack RIPK1, RIPK3, and either caspase-8 or FADD survived weaning and matured normally. RIPK1 functions in vitro to limit caspase-8-dependent, TNFR-induced apoptosis, and animals lacking RIPK1, RIPK3, and TNFR1 survive to adulthood. The role of RIPK3 in promoting lethality in ripk1−/− mice suggests that RIPK3 activation is inhibited by RIPK1 postbirth. Whereas TNFR-induced RIPK3-dependent necroptosis requires RIPK1, cells lacking RIPK1 were sensitized to necroptosis triggered by poly I:C or interferons. Disruption of TLR (TRIF) or type I interferon (IFNAR) signaling delayed lethality in ripk1−/−tnfr1−/− mice. These results clarify the complex roles for RIPK1 in postnatal life and provide insights into the regulation of FADD-caspase-8 and RIPK3-MLKL signaling by RIPK1.
原文链接:http://www.cell.com/cell/abstract/S0092-8674(14)00536-4
RIPK1 Regulates RIPK3 -MLKL-Driven Systemic Inflammation and Emergency Hematopoiesis
Summary Upon ligand binding, RIPK1 is recruited to tumor necrosis factor receptor superfamily (TNFRSF) and Toll-like receptor (TLR) complexes promoting prosurvival and inflammatory signaling. RIPK1 also directly regulates caspase-8-mediated apoptosis or, if caspase-8 activity is blocked, RIPK3-MLKL-dependent necroptosis. We show that C57BL/6 Ripk1−/− mice die at birth of systemic inflammation that was not transferable by the hematopoietic compartment. However, Ripk1−/− progenitors failed to engraft lethally irradiated hosts properly. Blocking TNF reversed this defect in emergency hematopoiesis but, surprisingly, Tnfr1 deficiency did not prevent inflammation in Ripk1−/− neonates. Deletion of Ripk3 or Mlkl, but not Casp8, prevented extracellular release of the necroptotic DAMP, IL-33, and reduced Myd88-dependent inflammation. Reduced inflammation in the Ripk1−/−Ripk3−/−, Ripk1−/−Mlkl−/−, and Ripk1−/−Myd88−/− mice prevented neonatal lethality, but only Ripk1−/−Ripk3−/−Casp8−/− mice survived past weaning. These results reveal a key function for RIPK1 in inhibiting necroptosis and, thereby, a role in limiting, not only promoting, inflammation.
原文链接:http://www.sciencedirect.com/science/article/pii/S0092867414005376
