

成都生物所王飞团队在基于合成致死策略靶向治疗KRAS突变型非小细胞肺癌方面取得新突破
来源:生物资源利用中心
作者:盛钰雯
时间:2025-04-25
近日,中国科学院成都生物研究所王飞团队在国际权威学术期刊Advanced Science(影响因子14.3,一区TOP期刊)上发表题为“Synthetic Lethality of SHP2 and XIAP Suppresses Proliferation and Metastasis in KRAS‐mutant Nonsmall Cell Lung Cancer”的研究论文,首次报道了来源于药用植物白花酸藤果中的天然化合物蒽贝素(Embelin)同时靶向SHP2和XIAP发挥合成致死效应抑制KRAS突变型非小细胞肺癌(NSCLC)的增殖和转移,为KRAS突变型肺癌的靶向治疗提供了全新的治疗策略和临床前证据。
大鼠肉瘤病毒癌基因同源物(rat sarcoma viral oncogene homolog,RAS)超家族是一个小 GTP 酶蛋白质超家族,由HRAS、NRAS和KRAS组成,被确定为癌症中最重要的癌基因家族之一。在所有RAS基因中,KRAS是突变最为频繁的,约占所有RAS突变的80%。KRAS突变具有显著的组织特异性,在结直肠癌(43%)、胰腺导管腺癌(85%)和非小细胞肺癌(NSCLC,31%)中尤为常见。作为一种分子开关,RAS 通过其非活性状态(GDP-RAS)和活性状态(GTP-RAS)之间的双向转换调节许多信号级联反应和生物进程。然而,RAS 突变可导致三磷酸鸟苷水解酶(Guanosine triphosphate hydrolase,GTPase)活性受损,促进活性 GTP-RAS 的生成和下游信号通路的持续激活,从而导致肿瘤细胞快速增殖,恶性程度增加。因此,抑制 KRAS 蛋白及其上下游通路可能是治疗癌症的有效策略。尽管KRAS长期被认为是“不可靶向”的癌基因,但随着共价抑制剂如sotorasib和adagrasib的问世,KRAS靶向治疗取得突破,然而快速出现的耐药性和疗效持续性差等问题仍严重限制了其临床应用。此外,由于毒性限制,联合免疫检查点抑制剂或其他靶向药物的治疗策略受到制约,阻碍了更具前景的“组合疗法”策略的开展。因此,迫切需要寻找新的策略治疗 KRAS 突变的相关疾病。
SHP2(Src homology domain 2 containing tyrosine phosphatase 2)作为受体酪氨酸激酶(RTK)下游信号复合体的关键成员,在多个RTK诱导的RAS激活通路中居中枢地位,其抑制可较广泛地克服KRAS抑制后产生的适应性耐药,同时增强KRAS-G12C抑制剂的疗效。除细胞内信号抑制外,SHP2阻断还能调节肿瘤微环境,如耗竭促肿瘤M2型巨噬细胞、促进M1型巨噬细胞极化,进一步发挥抗肿瘤效应。SHP2一度被认为是不可成药靶点,直到2016年,诺华公布了首个针对SHP2非活性构象界面口袋(N-SH2、C-SH2与PTP结构域之间的缝隙)变构抑制剂的研发进展,将研究重点转向SHP2的变构模式,SHP2抑制剂研发迎来突破。目前全球共有35款SHP2抑制剂在研发,其中已有17款SHP2变构抑制剂处于临床试验,绝大多数尚处早期的一期临床试验。加科思自主研发的JAB-3312是全球研发进展最快、也是唯一进入三期临床试验的SHP2抑制剂,与KRAS G12C抑制剂联合用药治疗NSCLC。然而,这些SHP2变构抑制剂的作用位点高度同质化,均靶向SHP2非活性构象界面口袋,且存在难以实现特异性抑制、细胞膜通透性差、生物利用度低等药代动力学障碍,同时不同肿瘤亚型对其敏感性存在异质性,耐药机制亦尚不明确,严重制约了其临床转化和广泛应用。合成致死(Synthetic Lethality)是指当两个基因同时失活时细胞死亡,而单独失活其中一个基因细胞仍可存活的现象。例如,PARP(聚ADP-核糖聚合酶)参与DNA单链损伤修复,而BRCA1/2基因突变会削弱同源重组修复(HR)能力。抑制PARP会导致BRCA突变癌细胞因DNA修复通路双重缺陷而死亡,而正常细胞因保留HR功能不受影响,该策略已在卵巢癌、乳腺癌等治疗中取得显著成功。因此,发现新的SHP2合成致死伴侣将为增强SHP2变构抑制剂的临床疗效,靶向如KRAS这类传统难以成药的癌基因提供了新的治疗思路。
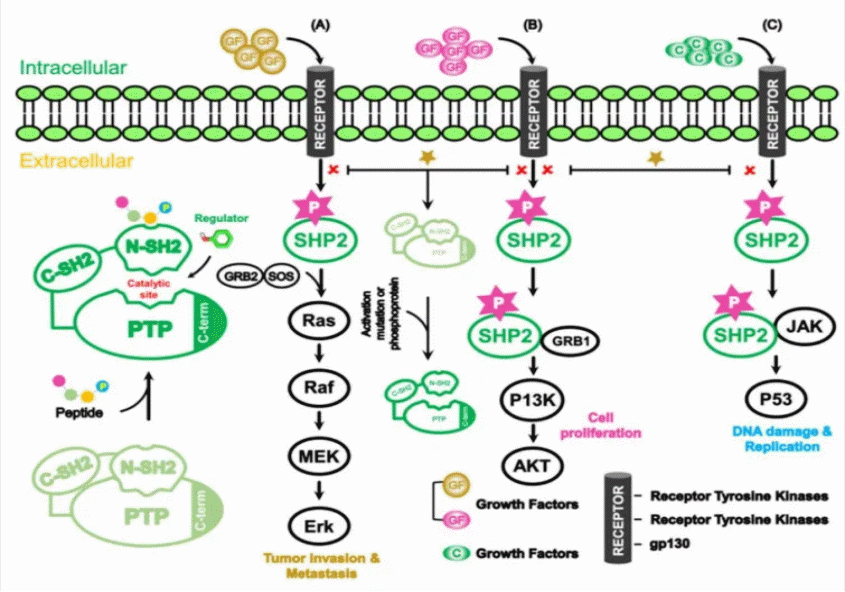
图. 蛋白酪氨酸磷酸酶(SHP2)参与多项信号通路
王飞研究员团队首次发现与目前广泛靶向SHP2非活性构象界面口袋的变构抑制剂如SHP099不同,蒽贝素能够与SHP2的PTP催化结构域中的关键残基Lys325特异性结合,实现对SHP2的特异性抑制,而对SHP1、PTP1B等磷酸酶活性没有影响。这一发现挑战了传统认为SHP2的磷酸酶结构域缺乏变构位点的观点,为开发具更高选择性、降低副作用的SHP2抑制剂提供了全新思路。进一步研究表明,SHP2和抗凋亡蛋白XIAP是潜在的合成致死伙伴,蒽贝素通过同时靶向SHP2和XIAP,显著抑制非小细胞肺癌细胞的增殖与迁移,诱导细胞衰老和内源性凋亡,并有效抑制MAPK、PI3K/AKT、JAK/STAT等多条致癌信号通路的激活。同时,蒽贝素破坏在RAS信号传导中发挥关键作用的SHP2/SOS1/Grb2信号复合体的形成,调节MIG-6和SPRY2克服MAPK信号通路的负反馈调节,从而发挥对KRAS突变型NSCLC的治疗作用。这些研究结果首次揭示了SHP2和XIAP是潜在的合成致死伴侣。与目前临床开发的靶向SHP2非活性构象界面口袋的变构抑制剂相比,蒽贝素具备更强的抑癌活性和更有效的负反馈抑制,为开发新型SHP2/XIAP双靶点抑制剂提供了骨架,也为克服KRAS突变型NSCLC中的凋亡耐受性和适应性耐药问题提供了新的治疗策略。
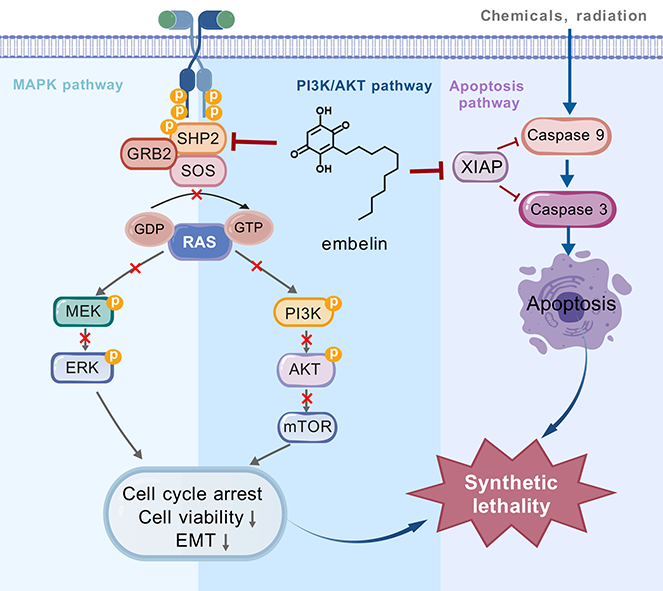
图. 蒽贝素作用机制示意图
王飞研究员为本论文的通讯作者;成都生物所博士生付乃洁、特别研究助理盛钰雯为本文并列第一作者。该研究得到了国家自然科学基金、中国科学院战略生物资源计划和中国科学院成都生物研究所前沿交叉项目等的支持。
