

开发植物药典、寻找特效天然药物
来源:Plant Cell
作者:Jennifer Wisecaver等
时间:2017-04-27
植物能释放成千上万种小分子化合物(也称次生代谢物)用以抵抗捕食者。你能说出几个?可卡因(cocaine)、尼古丁(nicotine)、辣椒素(capsaicin)
植物次生代谢物都是极其宝贵的处方药宝库,有些,已经被制成药物改善疾病治疗;有些,具有成瘾性使人类为它疯狂。不幸的是,即便科学家们能够比你多知道几种,也很难确定植物生产这些化合物的整个基因网络。了解植物次生代谢产物的生产机制,对改善医学治疗可谓是功勋一件。
4月14日发表在《Plant Cell》杂志的一篇文章,为鉴定这个庞大的植物基因网络提供了一个有效、有力的新方法。
“植物所合成的生物产品种类繁多。这项研究革命性地揭示了这些生物产品的天然属性,并且了解了它们的合成途径,”国家科学基金生物科学董事会的项目负责人Anne Sylvester、也是这项研究的资助人说道。
新方法建立在实验的基础上。
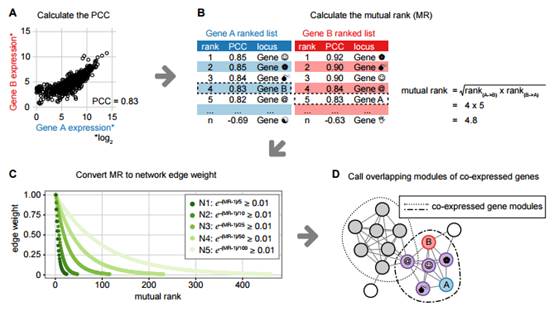
“我们假设在特殊化合物生产的基因网络调控中,面对同一种环境条件,机体的反应应该是相似的,”研究指挥人博后 Jennifer Wisecaver说。
为了验证这种假设,Wisecaver与另外两位研究人员(生命科学教授Antonis Rokas和本科生Alexander Borowsky)对8种模式植物的22000多个基因在不同条件下的调控反应展开了大规模数据分析。
“这项研究借助了范德堡先进计算中心的超级计算机,检测分析了植物出于各种特殊条件(如高盐、干旱、各种捕食者、病原体)时,体内的基因开合,” Wisecaver说。
与成千上万种基因分析相比,千种环境下的样本采集工作仅是小巫见大巫。因此,范德堡大学的科学家们专门为此设计了一个强大的算法,来克服这项困难。这种算法有能力从整个基因网络中鉴定由始至终表现相同的基因。
Vered Tzin(班古里昂大学,以色列) 和Georg Jander(康奈尔大学,美国)负责玉米的基因分析; Daniel Kliebenstein(加州大学戴维斯分校,美国)负责另一种模式生物拟南芥。
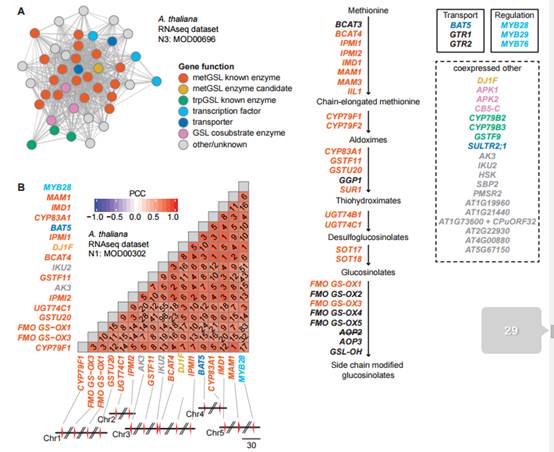
数据处理涉及数十个,甚至上百个小分子代谢物产生的基因途径,其中也包含了前期实验已经鉴定的部分。
之前的理论认为,产生这些次生代谢物的基因聚类存在于植物的基因组中。“这类想法都来自科学家们对细菌和真菌基因组的观察,它们的调控特殊代谢产物途径的基因都聚集在一起,” Rokas说。“但是植物的这类基因却大多分散在整个基因组中。因此,植物基因途径的研究策略需要与细菌和真菌区别开来。”
该篇《Plant Cell》向我们展示了一种新颖的、丰富的、很大程度上从未被使用过的高通量植物天然产物的遗传学基础和架构研究手段。
倘若这种方法能够经受住更广泛的研究验证,那么,它就是人类开启新型植物疗法闸门的关键开关。(来源:生物通 欧阳沐)
A global co-expression network approach for connecting genes to specialized metabolic pathways in plants
Abstract Plants produce a tremendous diversity of specialized metabolites (SMs) to interact with and manage their environment. A major challenge hindering efforts to tap this seemingly boundless source of pharmacopeia is the identification of SM pathways and their constituent genes. Given the well-established observation that the genes comprising a SM pathway are co-regulated in response to specific environmental conditions, we hypothesized that genes from a given SM pathway would form tight associations (modules) with each other in gene co-expression networks, facilitating their identification. To evaluate this hypothesis, we used 10 global co-expression datasets - each a meta-analysis of hundreds to thousands of expression experiments - across eight plant model organisms to identify hundreds of modules of co-expressed genes for each species. In support of our hypothesis, 15.3-52.6% of modules contained two or more known SM biosynthetic genes (e.g., cytochrome P450s, terpene synthases, and chalcone synthases), and module genes were enriched in SM functions (e.g., glucoside and flavonoid biosynthesis). Moreover, modules recovered many experimentally validated SM pathways in these plants, including all six known to form biosynthetic gene clusters (BGCs). In contrast, genes predicted based on physical proximity on a chromosome to form plant BGCs were no more co-expressed than the null distribution for neighboring genes. These results not only suggest that most predicted plant BGCs do not represent genuine SM pathways but also argue that BGCs are unlikely to be a hallmark of plant specialized metabolism. We submit that global gene co-expression is a rich, but largely untapped, data source for discovering the genetic basis and architecture of plant natural products, which can be applied even without knowledge of the genome sequence.
原文链接:http://www.plantcell.org/content/early/2017/04/13/tpc.17.00009.full.pdf+html?sid=4c8932c3-045f-4bd3-aee8-561b1f464409
